
To understand how big drug companies abuse the U.S. patent system, get a basic understanding of the U.S. patent system.
Patents encourage inventors to make their discoveries public by granting them a 20-year monopoly in exchange for filing a patent that describes their invention. A patent essentially grants a monopoly on prices and sales because no one else can sell something that uses the information that is covered by the patent. The length of a patent monopoly is always 20 years, regardless of whether the inventor invested years or millions of dollars, or if an invention came from a flash of insight. But the patent also ensures good ideas don’t remain secret. When a patent expires, anyone can use the valuable information in the patent or build on that invention.
To be granted a patent, patent applicants are supposed to show their invention is new and useful. To keep competitors off the market, drug companies file dozens, and sometimes even hundreds, of patent applications for the same drug. Patent examiners have limited time to review each one and sometimes grant a patent for obvious and not-new changes to a drug. Studies have shown that more than two-thirds of secondary drug patents are invalidated, meaning they should have never been granted patent exclusivity.
In 2021, there were an average of 74 approved patents on each of America’s ten top selling drugs. A drug company that wants to sell a competing generic drug would have to successfully challenge each of those 74 patents before they could offer their own version of the medicine. These challenges take years and millions of dollars. The prospect of high litigation costs and long delay in some case deter competitors from even trying to enter the market with a generic or biosimilar. The result: no competition and drug prices remain high.
Patents are intended to expire so others can build on those innovations to the benefit of the public. Ending a patent monopoly enables better competition, lowers prices, and gives anyone a chance to build on earlier innovation. But when the system is abused and patents simply extend monopolies, no one wins except the profiteering patent holder.
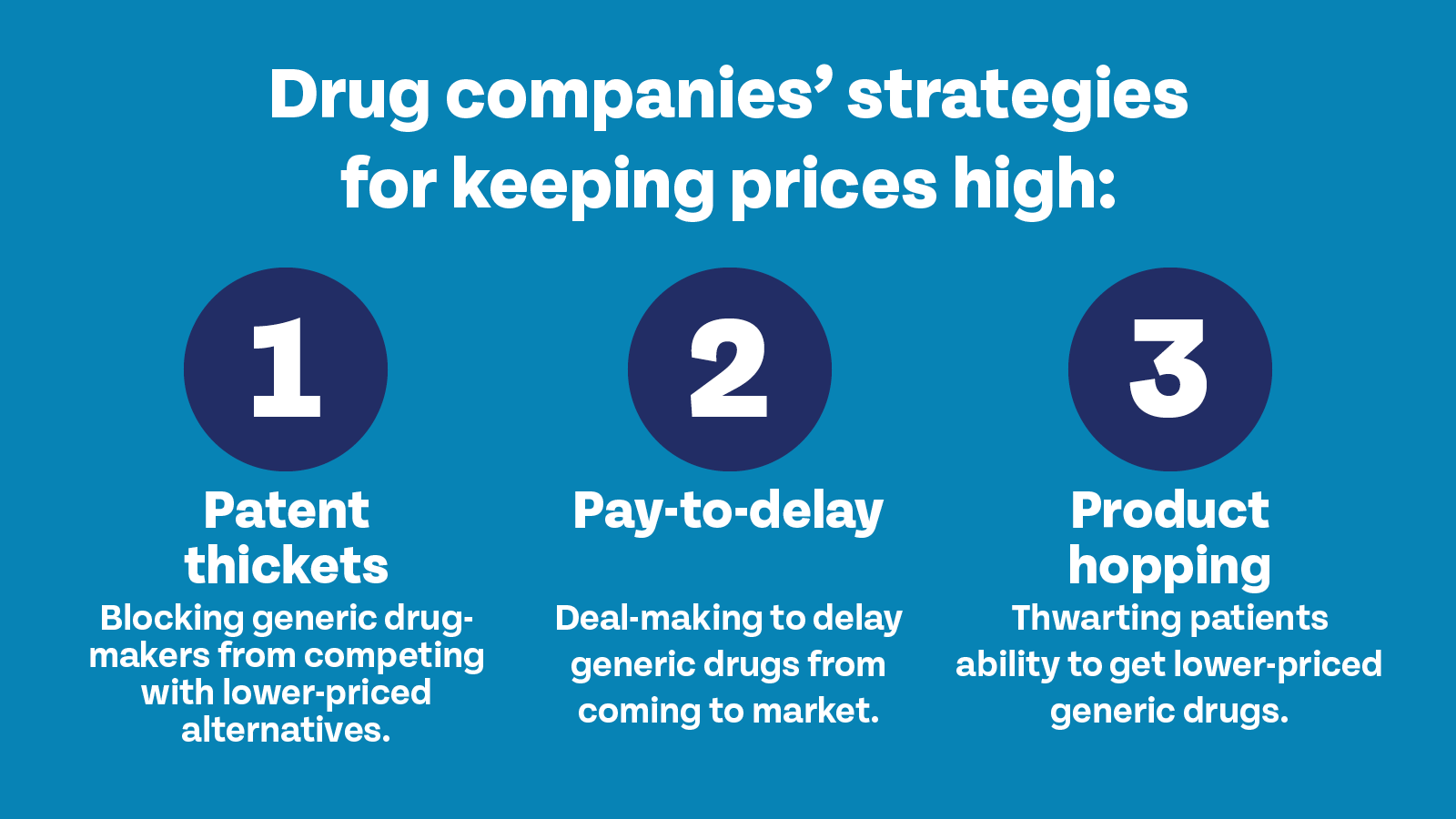
A patent thicket is created when a drug company strategically applies for and amasses dozens of approved patents on a single drug – essentially wrapping their drug in so many patents that no competitor could ever challenge them all.
For a generic drug competitor to be able to sell its lower priced version of a drug to patients, it must show that the brand-name drug company’s existing patents are invalid or don’t apply to their medication. The patents can be challenged at the U.S. Patent and Trademark Office or in federal court. But it takes years and millions of dollars to challenge a patent thicket.
These patent thickets extend the drug company’s monopoly by creating major hurdles for potential generic competitors, and ultimately cost patients and the public billions of dollars by delaying competition.
Patent thickets are made up of primary patents and secondary patents.
Primary patents typically cover a drug’s active ingredient or chemical composition – the main reason it is considered a “new” drug and the innovation which makes the drug eligible for a patent allowing monopoly pricing.
Secondary patents may cover any number of features of a medication that may or may not be a significant innovation. Examples include things like chemical alterations, manufacturing processes, safety practices, storage requirements, or “methods of use” – how to use the drug to treat specific conditions.
Patent thickets extend the 20 year monopoly.
Each patent carries its own 20-year monopoly. Drug companies often file applications for secondary patents years after the primary patent is granted. This allows drug companies to prevent competition for years or even decades past the end of a drug’s primary patents. Of the top 10 selling drugs, 66% of patent applications were filed after the Food and Drug Administration (FDA) approved the drug.
Patent thickets mean higher costs for patients.
Delays in generic competition caused by patent thickets cost patients and the public billions of dollars. For just three of the top selling U.S. drugs, Americans will spend an estimated $167 billion on brand-name drugs after generic competition begins in the European Union.
For more on how patent thickets work, read The Cost of Prescription Drug Patent Abuse report here.
Here’s an example of a patent thicket on an important cancer drug, preventing competition and keep prices high.
Revlimid is a drug used to treat multiple myeloma, a cancer of the blood affecting an estimated 35,000 people in the U.S., and other forms of cancer.
In 1998, drug company Celgene, now a subsidiary of Bristol-Myers Squibb, won FDA approval to sell thalidomide, a decades-old drug, to treat leprosy. A few years later, in 2005, Celgene won FDA approval to sell Revlimid, a slightly chemically altered version of thalidomide.
Celgene’s primary patent for the active ingredient in Revlimid was filed in 1996. Because patents last 20 years, that patent expired in 2019. However to maintain its monopoly pricing advantage over the years since it’s first patent, the drug company continued to file an additional 206 U.S. patent applications for Revlimid, 117 of which have been approved. As a result, Revlimid faces only limited generic competition now, and won’t face unrestricted competition until 2026.
Celgene has charged inflated monopoly prices for years. Celgene raised the price of Revlimid 23 times between 2005 and 2020. The price of a monthly supply of Revlimid has gone from roughly $6,000 in 2006 to $24,000 in 2022. Experts have estimated that Celgene’s extension of its Revlimid monopoly will cost Americans $45 billion.
For the full story of Revlimid, read The Cost of Prescription Drug Patent Abuse report here.
In a pay-for-delay deal, a brand-name drug company pays off a would-be competitor to delay it from selling a generic version of the drug. Without competition, the brand-name company can continue demanding inflated monopoly prices. And patients lose out on the savings they could have had, if the generic drug had been made available earlier.
The first generic competitor to apply to the FDA to sell a generic version of a drug gets their own period of exclusivity. The FDA won’t approve another generic version of the drug until 180 days after the first competitor starts selling its product.
To keep their prices high, and extend their monopoly, a brand-name drug company makes a pay-for-delay deal to get that first generic competitor to delay selling their equivalent drug. That way they can keep all generic competitors from pharmacy shelves.
The pay-off comes in many forms: outright payments of cash, in-kind gifts of free brand-name drugs (which the generic company can then sell for pure profit), and other valuable agreements. For example, the brand company might let a generic company start selling their competing drugs in some markets (like Europe) even before their patent expires.
Both drug companies win in a pay-for-delay scheme, but patients and the public lose. A single generic competitor can lower prices by 30% and competition from five competitors can lower prices by more than 80%.
For more on how pay-for-delay works, read The Cost of Prescription Drug Patent Abuse report here.
Lidoderm is a topical patch used to relieve pain associated with a complication from shingles, and Lidoderm was the preferred treatment in 2012 when a generic competitor was poised to start selling its own version of the drug. The generic drug company Watson challenged the Lidoderm patents so it could bring its drug to patients before Endo’s patent expired in 2015.
Endo Pharmaceuticals, the manufacturer of Lidoderm didn’t want to share its lucrative market with anyone. It’s profits from the drug totaled $948 million in 2012 alone.
So the two companies made a pay-for-delay deal in which competitor Watson agreed to abandon its patent challenge and to delay selling its generic drug by more than a year, from May 2012 to September 2013. In exchange, Endo compensated Watson in the following ways valued, according to the FTC, at approximately $250 million:
First, Endo agreed not to compete with Watson by selling its own “authorized” generic version of Lidoderm for up to 7½ months. The Federal Trade Commission estimated Endo’s promise to withhold its own authorized generic would allow Watson to earn at least an additional $214 million during its first six months on the market as the sole generic producer.
Second, Endo agreed to give Watson free branded Lidoderm patches, valued at over $90 million, which it could sell on the monopoly-priced market. Endo also agreed to give Watson up to an additional $144 million of branded Lidoderm if the FDA did not approve Watson’s generic application.
Both companies profited and patients had to wait longer for the cost-savings they could have gained if other generic versions had come to market earlier.
For the full story of Lidoderm, read The Cost of Prescription Drug Patent Abuse report here.
Product hopping is a tactic used by brand-name drug companies to keep patients from getting competing lower-priced generic drugs at the pharmacy counter.
In every state, once a generic drug is approved by the Food and Drug Administration as equivalent to the brand name drug, a pharmacists can substitute the generic drug for the prescribed brand-name drug. These substitution laws help patients easily save billions of dollars each year.
Drug companies use product hopping when their patents are close to expiring. To keep their monopoly-pricing, the company alters the patented drug just enough that the available generic versions cannot be automatically substituted at the pharmacy counter. Brand-name drug companies then encourage doctors to write new prescriptions to move their patients to the new version of the original medication. The pharmacist is now prevented from helping the patient get a lower priced drug because they can’t automatically substitute the lower priced generic drug at the pharmacy counter.
Sometimes, to be sure that all doctors move their patients to the newer version of the same drug, the company will remove the original version from the market entirely before the generics become available. Doctors have no choice but to write a new prescription for patients and when the generic is on the pharmacy shelves, it is no longer allowed to be substituted for the “new” prescription.
In this way, product hopping allows a brand-name drug company to thwart meaningful competition even when a lower priced generic version of a drug is available. The drug company continues to dominate the market with inflated monopoly prices while most patients don’t get the lower price and greater value of an available generic drug.
For more on how product hopping works, read The Cost of Prescription Drug Patent Abuse report here.
Suboxone is a treatment for opioid addiction, a major scourge in communities across the country. Importantly, when introduced, it was the only treatment approved for at-home use, which meant easier access to treatment for those struggling with opioid addiction.
Several years before its drug monopoly exclusivity was set to expire, executives at Suboxone manufacturer Reckitt Benckiser Group (“Reckitt) switched the form of its drug from a tablet to a thin film that dissolves under the tongue. The drug itself was the same.
Then it went about getting doctors to move their patients to the new film version of the treatment. They did it in two ways.
First, they began a fear campaign saying children might more easily misuse the tablet and that the individually wrapped child-proof packaging of the film was safer. The Food and Drug Administration rejected their safety claims saying its clinical trial was poorly designed and “not useful for demonstrating any difference in the safety profile or abuse potential of these two formulations [tablet and film].”
Second, Reckitt eventually forced doctors to prescribe its film by completely removing its tablet from pharmacy shelves.
Because generic versions of the drug, in tablet form, could not be automatically substituted for the brand-name drug, newly available in dissolvable film form, Reckitt delayed significant competition by years, and ultimately maintained an 80% market share after generic competition began.
The result of the product hopping tactic means many patients who needed the opioid treatment were preventing from buying the lower priced generic tablets from other companies—and enjoying substantial cost-savings.
For the full story of Suboxone, read The Cost of Prescription Drug Patent Abuse report here.
The story of how one drug company, AbbVie, used all three tactics (patent thickets, pay-for-delay, and product hopping to extend monopoly prices for the world’s best-selling drug, Humira.
Humira, the brand-name of the drug adalimumab, is a treatment for rheumatoid arthritis, psoriasis, Crohn’s disease, and more – conditions affecting over 10 million patients in the U.S.. Because it is a “biological” drug, Humira faces competition not from generic versions of the drug, but from so-called “biosimilar” drugs.
First approved by the FDA in 2002, Humira’s primary patents ended seven years ago in 2016. Yet, because AbbVie, the maker of Humira, has focused less on innovating new drugs but more on maintaining monopoly pricing of its profit-maker Humira, the drug did not face any biosimilar competition in the U.S. until early 2023.
Abusing the patent system is a business strategy that has worked well for AbbVie and its shareholders. But patients, health plans and government drug programs have borne the cost of its patent abuse.
Humira is a quintessential “blockbuster” drug for AbbVie. In 2021, Humira was the top selling drug in the U.S., bringing in $17.3 billion. Between 2003 and 2021, AbbVie raised the drug’s price 27 times. In 2021, a year’s supply of Humira cost $77,586.
AbbVie has built a massive patent thicket. It applied for 312 patents for Humira, 166 of which have been granted. AbbVie used its patent thicket to block or delay all U.S. competition until early 2023. Competition in the U.S. may continue to be restricted until Humira’s last existing patent expires in 2037. And the wait for unrestricted competition could be even longer: AbbVie has four additional patent applications pending before the U.S. patent office, potentially extending its patent thicket further into the future.
A 2021 Congressional investigative report showed how AbbVie used its patent thicket in a pay-for-delay deal which extended monopoly pricing in the U.S. by allowing competitors to sell their products earlier in Europe.
Maintaining its patent protection in the U.S., AbbVie enticed biosimilar drug competitors with a deal they couldn’t refuse: early entry into the European market. For 6 of the 9 drug companies that settled with AbbVie to begin biosimilar sales in the U.S. in 2023, their agreement allowed them to begin sales in the European Union in 2018. The deal delayed U.S. competition 6 years beyond its initial expectation
AbbVie introduced a higher concentration version of Humira in July 2018. It claimed to be introducing the higher concentration because it is less painful for patients. But internal company documents showed that the move was part of its biosimilar “defense strategy,” meaning it was a change that would help the company keep patients paying its monopoly prices.
AbbVie obtained FDA approval for the higher concentration version of Humira in 2015, but waited three years to begin selling it. The delay ensured that its competitors had invested significantly in creating biosimilar drugs that could be substituted for the original Humira concentration.
Doctors were encouraged to write prescriptions for the newer concentrations – the “product hop.” AbbVie can keep most of its patients on their higher priced drug because only one biosimilar competitor has achieved the “interchangeable” status that will allow pharmacists to automatically replace it for Humira. When patients lose out on automatic pharmacy substitution because of the product hop, it makes it more difficult for them to get the numerous biosimilar competitors that will become available throughout 2023.
These three tactics have successfully delayed patient access to biosimilar competitors, at a huge cost: $19 billion.
We need significant patent reform to allow patients swifter access to the lower prices of generic and biosimilar drugs. Patients, insured families and our government health plans will save money from the resulting price competition.
Federal legislation will help regulators stamp out specific anti-competitive practices and give them additional power to identify emerging tactics that block generic and biosimilar drugs from coming to market.
Some important PTO reforms include:
Less emphasis on swift review of patent applications and more emphasis on quality review. We urge a return to the mission of the PTO to serve the public. It is time to shift away from an overemphasis on serving patent applicant “clients” by reviewing applications too swiftly. The public mission requires high quality examination of drug patent applications, with the collaboration of experts from the Food and Drug Administration (FDA), to prevent approvals of weak, duplicative or anti-competitive patent applications. Patent examiners should prioritize their work to avoid approving patents filed for the purpose of creating patent thickets and other abusive tactics that prevent or postpone generic competition.
More stringent review of patent applications for prescription drugs already on the market. Examiners must reject patent applications that allow monopoly pricing without any clear substantial change to the medication or its efficacy. Patent applicants should clearly disclose when a new or continuation application claims aspects of or improvements to an existing drug already on the market. The agencies should flag PTO and FDA applications which correspond to substantially similar drugs, share information provided by applicants (especially regarding clinical test results and the necessity of clinical testing), and spend more time reviewing those applications for inaccuracies or outright fraudulent and deceptive claims. Patent examiners should receive extra support from FDA experts knowledgeable with that approved drug or drug class so they can assist the patent examiner in understanding whether the new patent meets the required tests for subject matter eligibility, usefulness, nonobvious and novelty.
Restore and protect the utility of the Patent Trial and Appeal Board (PTAB). This arm of the PTO offers an alternative to litigating patents in the federal court system. The PTAB provides a quicker and less expensive way to challenge the validity of patents and is the only opportunity for a member of the public to challenge the patentability of a claim in an approved patent. In recent years, internal changes have narrowed the opportunities to bring challenges to the PTAB. Improving the effectiveness of the PTAB and restoring the original purpose of this alternative to litigation could result in earlier market entry of generics and biosimilar drugs.
The agencies should establish regular information sharing and joint training with a collaborative approach to auditing, inspections and enforcement actions. Pharmaceutical business practices and strategies regularly employ tactics that unfairly manipulate the patent and drug application systems to hinder generic competition. Therefore, we need a coordinated approach to enforcement to leverage the limited resources of each agency to ensure regulatory and statutory compliance by drug companies.
Many health care policy solutions are proposed, analyzed and decided with little or no consumer and patient input. Despite the complexity of patent and drug approvals, the patient and consumer voice is still an essential part of policy deliberations. When decision makers lose touch with the end-user, in this case, the patient and consumer community, policy decisions sometimes unwittingly or even in some cases purposefully put the needs and interests of the consumer last. Consumers find it difficult and time-consuming to comply with the strict formal input opportunities offered by these agencies, such as filing regulatory comments. Monitoring and reading technical applications are difficult even for highly trained chemists and patent attorneys. Other health agencies can serve as a source of ideas for how to better involve consumers in health policy considerations. The FDA and PTO should identify and consider more creative ways to solicit input from the patient/consumer community, and involve those audiences in that exploration. Then the agencies, with input from the consumers/patients, should implement an effective patient engagement model with the goal of ensuring that generic and biosimilar drug competition is restored.
Tell President Biden to stop drug patent abuse so we can save money on generic drugs.



